
山西煤化所在金属间电子化物表面自发水解离促进CO₂甲烷化研究中取得重要进展
近日,中国科学院山西煤炭化学研究所温晓东团队在理论-实验-数据一体化研究思路指导下,针对金属间电子化合物(IE)催化CO₂甲烷化研究取得重要进展,研究成果以“Spontaneous water dissociation on intermetallic electride LaCu0.67Si1.33 enhances electrochemical methanization of CO2”为题发表在《Nature Communications》期刊上。
利用可再生电力驱动的二氧化碳(CO2)电还原转化为甲烷是一种可持续的减少对天然气依赖的方法。然而,这一过程目前受限于效率和耐久性不足,这主要源于现有催化剂上水解离(WD)和质子耦合电子转移之间的动力学差异。为了提高CO2电还原为甲烷的效率,研究者们致力于设计更高效的催化剂,特别是铜(Cu)基催化剂。尽管如此,如何通过催化剂设计来同步实现高效的水解离和CO2活化仍然是一个挑战。
团队基于自研的机器学习加速结构预测方法(J. Phys. Chem. C, 2020,124,17244−17254),结合基于密度泛函理论(DFT)的高通量计算,揭示了金属间电子化合物材料关键活性位的富电子结构,并进一步预测其具有优异的自发水解离能力。结合LaCu0.67Si1.33材料制备并设计表征实验,利用H2O-程序升温表面反应(H2O-TPSR)证实了在该金属间电子化合物表面发生了显著的自发水解离现象。在碱性流通池中,IE LaCu0.67Si1.33催化剂在相对于可逆氢电极(vs. RHE)−1.21 V的还原电位下表现出72%的甲烷法拉第效率(FE),并在−1.52 V vs. RHE下达到476.7 mA cm⁻2的甲烷偏电流密度。密度泛函理论计算进一步表明,在IE LaCu0.67Si1.33催化剂上,甲烷化反应路径在热力学/动力学上更为有利。
通过数据挖掘,结构表征及理论预测,团队揭示了IE LaCu0.67Si1.33催化剂独特的活性位富电子结构和自发水解离能力,为CO2的电化学甲烷化以及其他可能涉及水解离过程的可再生能源转化,提供了一个极具前景的催化剂体系。以上工作得到国家科技部重点研发计划(批准号:2022YFA1604100),国家自然科学基金杰出青年基金(批准号22225206),中国科学院前沿科学重点研究项目(ZDBS - LY - 7007)等项目的支持。
本成果由中国科学院山西煤炭化学研究所温晓东团队和北京大学骆明川团队合作完成。
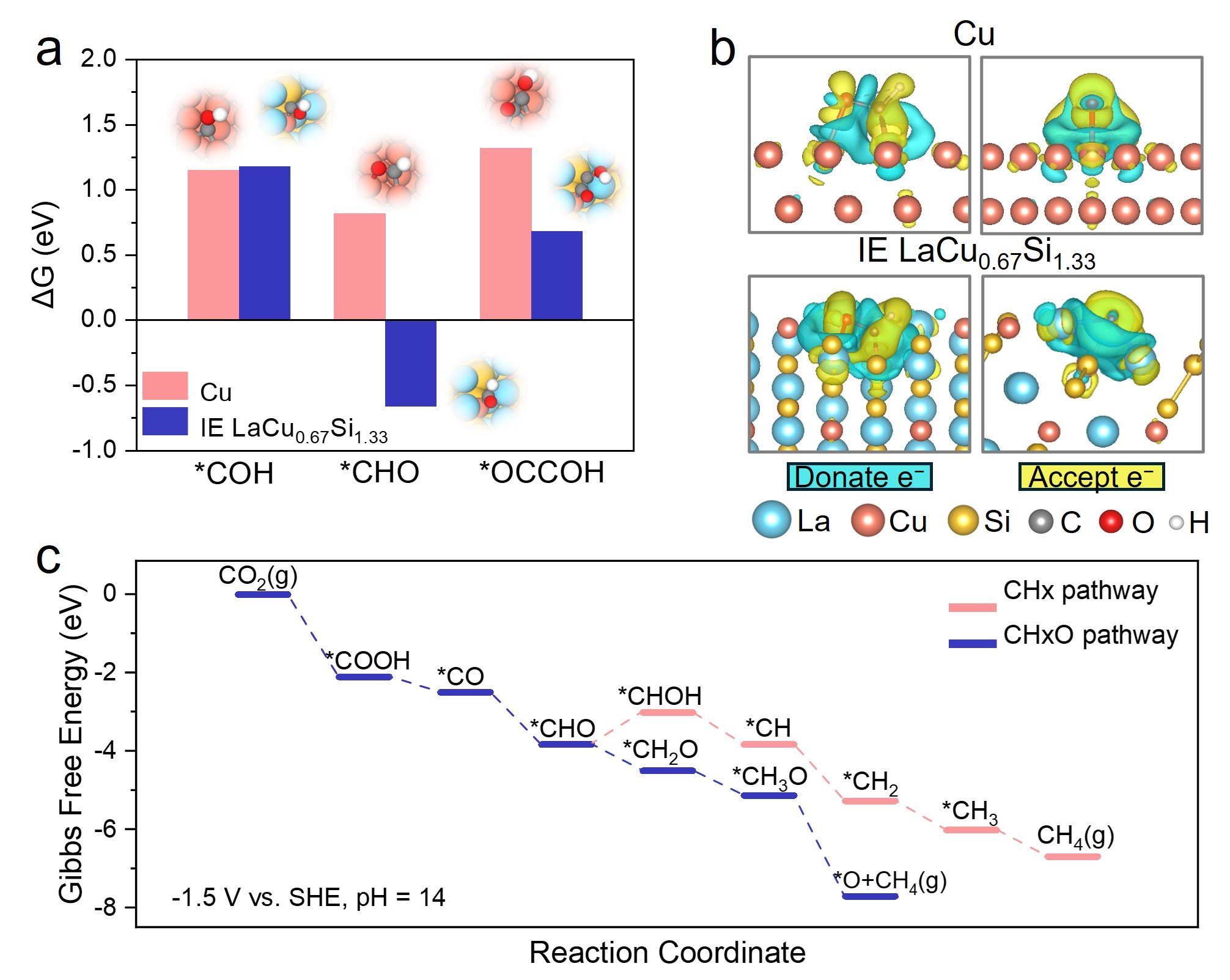
DFT计算(a) *CO反应系列中间物种在IE LaCu0.67Si1.33和Cu表面加氢和碳碳耦合反应的吉布斯自由能;(b)*CHO反应中间物种在IE LaCu0.67Si1.33和Cu表面的电荷密度差图;(c)CO2-CH4转变不同反应路径中间过程的吉布斯自由能。
(煤炭高效低碳利用全国重点实验室)
附件下载:



